
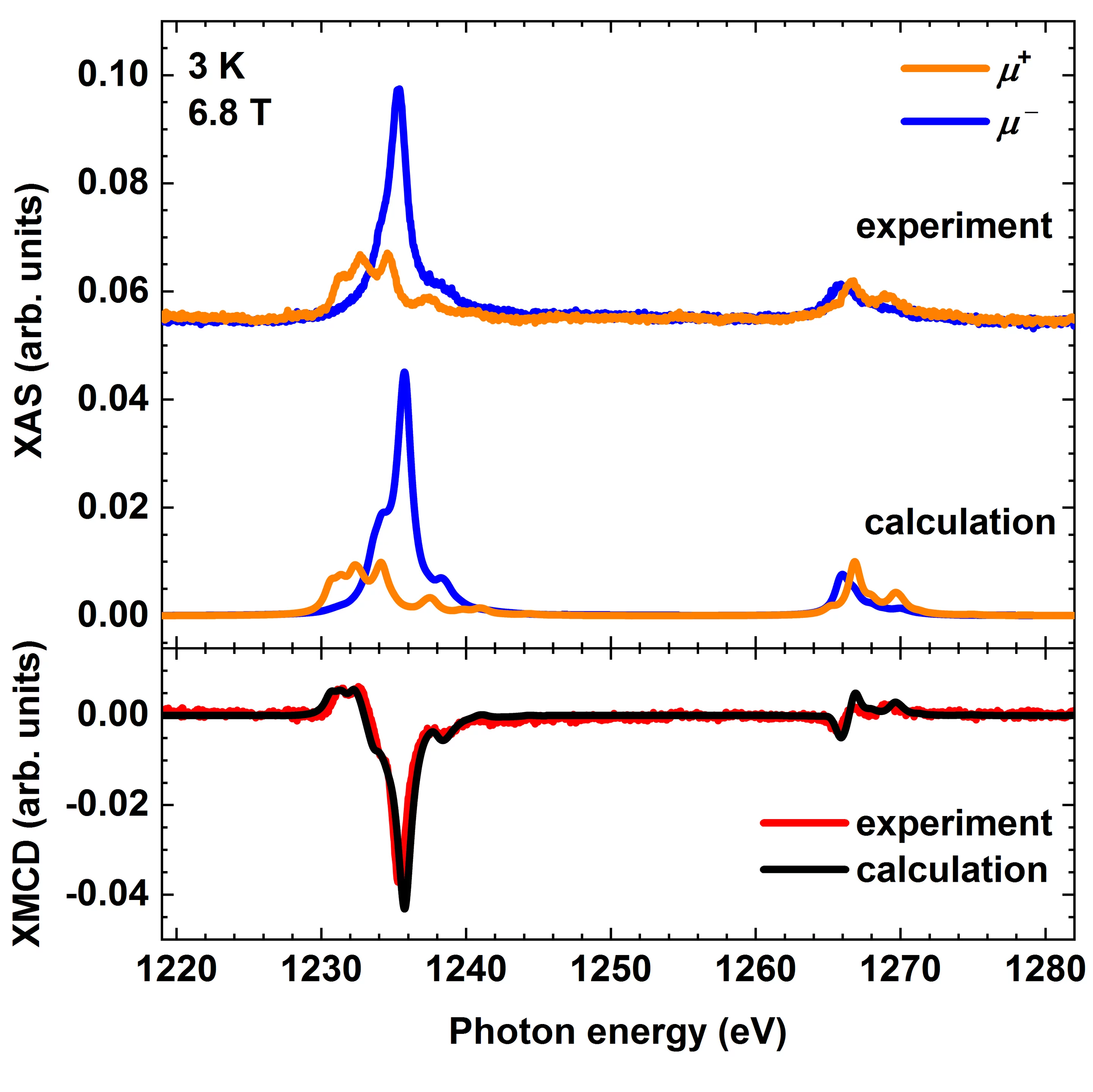
Theoretical calculations to reproduce X-ray absorption and X-ray magnetic circular dichroism (XMCD) spectra are important to learn about the electronic structure and magnetism of the experimental system on which the spectra were recorded. So far existing methods either rely on a large number of empirical inputs or they are computationally heavy. Now there is a novel method available which allows to calculate soft X-ray absorption and XMCD spectra only from structural input. The method called ligand-field density functional theory (LF-DFT) calculates the ligand-field parameters of 3d transition metal and lanthanide atoms using density-functional theory (DFT). It allows to reproduce correctly the atomic multiplets in the X-ray spectra while being independent on any empirical input. As an experimental test system the scientists have employed a monolayer of a terbium sandwich molecular complex on an oxide surface. The surface serves to orient the molecules, however, it provides only weak interaction with the molecules. The spectra calculated by the LF-DFT method are in excellent agreement with the experimental spectra measured at the X-Treme beam line of the Swiss Light Source.
Contact
Dr. Jan Dreiser
Staff Scientist
PSI, Photon Science Division
Forschungsstrasse 111, 5232 Villigen PSI, Switzerland
Telephone: +41 56 310 5895
E-mail: jan.dreiser@psi.ch
Original Publication
Non-empirical calculation of X-ray magnetic circular dichroism in lanthanide compounds.
Harry Ramanantoanina, et al.
Chemical Communications 55, 2988 (2019)
DOI: 10.1039/c8cc09321k