

Automating epitaxy for functional materials discovery
Pulsed laser deposition (PLD) is one of the few techniques capable of a highly controlled growth of epitaxial thin films of a broad range of compositions, including transition metal oxides, oxynitrides, halides and dichalcogenides, among others. Attaining single-crystal-like film properties (e.g., high crystallographic ordering, low defect density, accurate stoichiometry control) is a goal for many in the scientific community, as this would significantly enhance and advance the fundamental understanding of material properties in the context of new technological breakthroughs. The general limitation is the immense effort required to find optimal process parameters for growing single-crystal-like films, often proceeding through a time-consuming trial-and-error approach, whereby the parameter space is probed until optimized material figures of merit are found. As a result, more time is spent optimizing a new material composition than performing scientific experiments. Instrument automation can consequently be employed to enable the optimization of the growth parameters.
Recently, the Materials Software and Data (MSD) group (in the LMS lab), who develops the AiiDA workflow infrastructure for simulations, successfully demonstrated the extension of the platform to drive robotic experiments in the case of automated battery cycling (P. Kraus et al., J. Mater. Chem. A 12, 10773 (2024)), making the whole process ready to be integrated with machine-learning techniques, towards a fully autonomous optimization of the device. The adopted automation approach is very general and can be thus extended to many more experimental applications. In particular, combining automation techniques and machine learning with advanced epitaxy infrastructure and in situ monitoring tools available in the Thin Films and Interfaces (TFI) group at PSI, the process optimization can be fully automated to yield optimal growth parameters, ideally for material compositions never grown before. The automation of PLD is a topic
presently in its infancy, with two other research groups attempting to achieve a similar outcome. However, the in situ techniques available and the knowledge gained over time on the fundamentals of the PLD process in the TFI Group at PSI, combined with the experience in automation and simulations of the MSD group at PSI, give us a unique advantage in the successful realization of the proposed technology.
The project is a collaborative effort of the MSD group with Thin Films and Interfaces (TFI) group at PSI (Dr. Nikita Shepelin)
Contact: MSD group
FISH4DIET
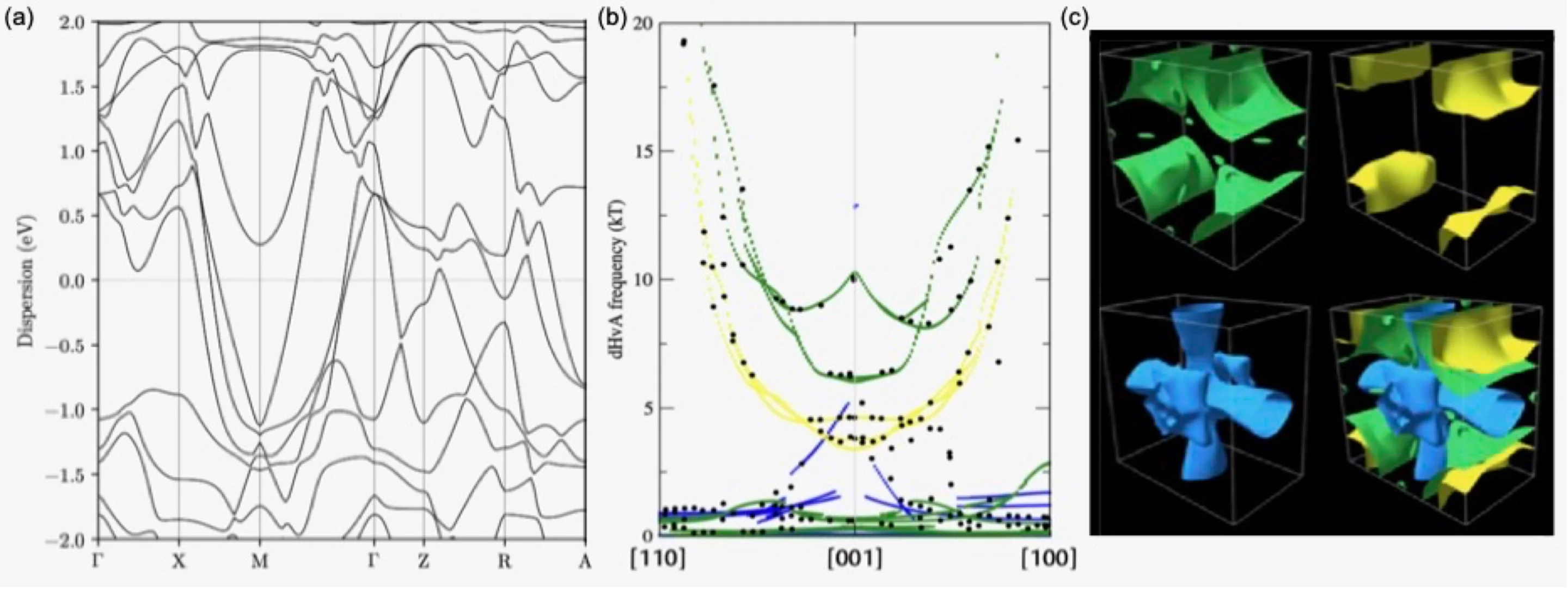
The Fermi surface (FS) of a solid separates occupied from unoccupied electron states at zero temperature. This concept, essential for understanding the behavior of electrons in solids, is also a very useful tool for property predictions of metals and doped semiconductors, particularly when combined with DFT simulations. However, to achieve a good accuracy of the calculated properties, a very dense sampling of the FS in reciprocal space is required, and direct first-principles calculations are limited by the computational time. To circumvent this difficulty, interpolation schemes have been developed, but either their practical utilization is far from trivial, or their accuracy is limited. Interpolation using Maximally Localised Wannier Functions (MLWFs) allows obtaining accurate results and approaches are being developed in order to automate the process.
One of the objectives of the project "Fermi Surfaces from High-throughput Ab Initio Calculations for the Discovery of Exceptional Topologies" (FiSH4DiET) is to develop very general tools to obtain efficiently accurate Fermi surfaces from high-throughput ab initio calculations, and to compute the related properties automatically.
With these tools, a database of FSs will be generated and made available openly. The latter will allow for the discovery of materials with exceptional topologies (and hence properties) of the FS, such as quasi-2D materials, tiny-pocket materials in the ultra-quantum regime, multi-valley systems and Kohn anomalies. In addition, in FISH4DIET we develop automated workflows to generate, test and verify accurate pseudopotentials, and deliver them in open libraries. Pseudopotentials are an essential ingredient of most density-functional-theory (DFT) simulations, especially when adopting a plane-wave basis set: having a curated, high-quality set of pseudopotentials is essential to have precise DFT results.
The project is a collaborative effort of the MSD group with Université Catholique de Louvain in Belgium (Prof. Gian-Marco Rignanese)
Contact: MSD group
Interoperability between the ETH Domain Repositories (IntER)
With this first-of-its-kind interoperability project, Eawag/LIB4RI, WSL and PSI aim to jointly define and implement a basic blueprint for the metadata interoperability to demonstrate the feasibility of achieving interoperability between data catalogues and repositories in the ETH Domain.
We aim to define a common understanding and an agreed compliance level with national and international metadata standards relevant to ETH Domain data catalogues and repositories, and subsequently take all required steps to implement and comply with them, in order to improve the visibility of datasets in the ETH domain and beyond.
This will include, as major deliverables, improving the link between datasets and scientific publications, and connecting the involved repositories with each other and with well-established central search portals. This project will also uncover hidden challenges and barriers to interoperability, paving the way for other repositories in the ETH domain to join our interoperability efforts.
The project is a collaborative effort of the MSD group with Eawag/Lib4RI (Dr. Fabian Felder), WSL/Envidat (Dr. Ionut Iosifescu Enescu) and the AWI department at PSI (Dr. Carlo Minotti).
Contact: MSD group
MARVEL Pillar 3: Digital Infrastructure of Open Simulations and Data
This is one of the four core pillars of the third phase of the NCCR MARVEL. The goal of this Pillar is to provide the critical digital infrastructure needed to enable materials design and discovery in the 21st century.
It will focus on 3 major objectives:
- prepare for the exascale challenge, both in terms of code performance and scalability on (pre-)exascale machines, and high-throughput performance to drive millions of individual HPC simulations;
- make all codes, algorithms and workflows widely usable in a simple and optimized way also by non-experts, via the services provided by the AiiDAlab infrastructure and the Quantum Mobile virtual machine; this will also ensure reproducibility of all research and facilitate FAIR access and sharing of the resulting data via the Materials Cloud portal;
- critically, deliver a self-sustaining long-term infrastructure for simulations for the whole Swiss materials-science research ecosystem (and beyond) also after the end of the NCCR, to deliver automated simulations as microservices, as a key component for autonomous materials discovery and characterization.
The project leaders of this pillar are Dr. Giovanni Pizzi (MSD group leader) and Dr. Joost VandeVondele (CSCS and ETHZ), and the principal investigators include Dr. Sara Bonella (CECAM and EPFL) and Prof. Michael Herbst (EPFL).
Contact: MSD group

MARVEL Pillar 4: Long-term Integration in the Swiss Scientific Landscape
This is one of the four core pillars of the third phase of the NCCR MARVEL. The goal of this Pillar is to plan, deploy, and ultimately phase-in the post-2026 embedding of MARVEL in the Swiss scientific landscape, in the form of core partnerships with PSI and Empa.
To reach these goals and objectives, two main efforts have to be coordinated: (i) development and implementation of reliable and robust open-source computational tools to understand, predict, and characterize experimental results, and (ii) their deployment as user friendly turn-key solutions. Resources are being invested in the creation of such tools, that boost cooperation between experiments and simulation. This enables high levels of standardization in how calculations for a specific experiment are performed and how results are analyzed and processed. Tools such as AiiDAlab are, in fact, specifically designed to enable experimentalists to independently access the submission of calculations and the analysis of the automatically processed data.
The project leaders of this pillar are Prof. Nicola Marzari (LMS laboratory head), Dr. Carlo Pignedoli (deputy group leader at Empa), Prof. Christian Rüegg (PSI director and SCD division head)
Contact: LMS laboratory
PREMISE
PREMISE (Open and Reproducible Materials Science Research) aims to establish, promote and facilitate the adoption of FAIR ORD practices in Materials Science, focusing on enabling the treatment, at the same level, of experimental and simulation data.
In PREMISE we develop metadata standards for machine-actionable interoperability between electronic lab notebooks (ELNs)/lab information management systems (LIMSs) and workflow management systems (WFMSs), and apply these standards to Materials Science ontologies. We then collect, design and disseminate best practices for generating ORD as a natural part of the research process, rather than as an additional duty for researchers. Our deliverables are demonstrated via concrete pilot use cases, chosen to be applicable to the broad field of Materials Science, and generalisable to other disciplines. We leverage two robust open platforms, developed and maintained within the ETH domain, compliant by design with FAIR requirements for experiments (openBIS) and simulations (AiiDA+AiiDAlab). We will bring them "to the next level" by implementing our novel set of ORD practices and demonstrating how an ELN/LIMS and a WFMS can be made seamlessly interoperable. We expect our deliverables to be essential components of the emerging field of autonomous laboratories, where automated simulations and robotic experiments are combined via artificial intelligence in closed feedback loops, ultimately accelerating materials discovery and characterisation.
The project is a collaborative effort of the MSD group with Empa (Nanotech@surfaces laboratory: Dr. Carlo Pignedoli, Materials for Energy Conversion: Prof. Corsin Battaglia) and ETHZ (Scientific IT Services: Dr. Bernd Rinn, Dr. Caterina Barillari, Dr. Henry Lütcke)
More information can be found on the PREMISE website.
Contact: MSD group

SwissTwins
The SwissTwins project aims at developing key technology for future HPC exascale services, spanning from virtual clusters, data streaming, containerisation, secure HPC access, workflows and more. The project is lead by the Swiss Supercomputer Center CSCS, and involves also other institutions: PSI, EPFL and ETHZ.
In particular, the MSD group at PSI leads work package 2, focusing on further development of the AiiDA workflow engine. On the one hand, the goal is to expand the support of AiiDA to further domains, with particular focus on the weather and climate (W&C) communities. On the other hand, the goal is to ensure compatibility and scalability on future HPC architectures, including support for REST API access to supercomputers (e.g. via the FirecREST API), containerised codes (e.g. via the SARUS technology), and integration with novel technologies such as virtual cluster technology, data streaming technologies and customised optimisation toolchains.
Contact: MSD group
SNSF project "Discovery, design, and characterization of novel Na-ion battery cathode materials"
In response to the escalating global energy demands driven by an expanding population and the imperative for climate neutrality, our society faces challenges that necessitate groundbreaking research across various disciplines, particularly in electrochemical energy storage. Advancing battery technology is pivotal in achieving a successful transition to a carbon-neutral society. The adoption of more renewable energy sources on the electrical grid and the electrification of transportation rely on innovative energy storage technologies that are both economically viable and efficient. While rechargeable lithium-ion batteries (LIBs) have revolutionized the energy-storage market and facilitated the widespread use of portable electronic devices and electric vehicles, their cost remains an obstacle due to limited Li resources. This cost challenge underscores the pressing need to explore alternative and cost-effective energy storage solutions. Sodium-ion batteries (SIBs) have emerged as a compelling option due to the greater abundance of Na resources compared to Li. Although SIBs exhibit a slightly lower energy density compared to their Li-ion counterparts, stemming from the larger atomic radius and marginally higher ionization potential of Na, their sustainability and cost-effectiveness, driven by the abundance of Na resources, positions them as very promising candidates for energy storage solutions. As the pursuit of renewable energy sources intensifies to combat climate change, the development and optimization of SIBs represent a crucial step towards a cleaner and more sustainable energy future.
Motivated by these challenges, this project is driven by the following objectives. Firstly, an extensive high-throughput screening of all known inorganic Na-containing materials will be conducted to discover novel SIBs cathode materials exhibiting exceptional electrochemical and thermodynamic properties, with the overarching ambition to surpass existing commercial LIBs. Concurrently, an in-depth characterization of Mn-Fe-Al-based layered oxides, renowned for their high energy density, reversible capacity, and cycling stability, will be performed. The specific focus is on identifying optimal concentrations of Mn, Fe, and Al that result in peak battery performance. This investigation relies on advanced first-principles simulations, conducted in close collaboration with experimental partners to synergize theoretical and empirical insights. Ultimately, the project aims to employ the technique of substitutional doping to design novel cathode materials with enhanced mechanical and electrochemical properties. This process starts with the most promising candidates identified through the high-throughput screening.
The project leader is Dr. Iurii Timrov (LMS/PSI).
Research Partnership Grant (RPG) with east and southeast Asia "Accurate and efficient electronic-structure functionals for spectra of materials: application to carbon quantum dots as photocatalysts for water treatment"
As the world continues to undergo rapid urbanization with an increasing human impact on the earth, humanity is facing major global environment and energy challenges. It is generally acknowledged that the development of advanced engineered materials is crucial to address the needs of energy, environment, and sustainability. Among others, the increasing accumulation of organic pollutants in water is a pressing problem, especially in developing countries where environmental regulatory and monitoring framework is not properly enforced. Efficient removal or degradation of these pollutants is of prime importance for a sustainable future. Among many contenders, photocatalytic technologies stand out due to the utilization of semiconductors and inexpensive sunlight for the removal of these pollutants. In a photocatalytic degradation, photo-generated electrons and holes are reactive species that can drive different redox reactions and transform an environmental pollutant into a benign or less harmful product. During recent years semiconducting carbon quantum dots (CQDs) have emerged as promising candidates for the detection of heavy metal ions in water and for an efficient photocatalytic degradation of aqueous pollutants.
In the quest for an increasingly better understanding and design of CQDs, ab-initio simulations of the spectral properties of CQDs are irreplaceable tools. However, while well-established ab initio approaches exist to access the spectral domain, those are either too computationally expensive (Green’s function theory) to address the complexity of a realistic system, or not accurate enough (time-dependent DFT) to accurately describe the spectra of complex materials. The main goal of this project is to overcome this limitation and study the electronic structure and the optical response of semiconducting carbon quantum dots (CQDs) in realiastic condition. To this end we will leverage the complementary and versatile experience of the partners in the development of advanced first-principles quantum-mechanical methods for materials simulations at the atomistic scale. In particular, we plan to combine a novel class of orbital-density-dependent spectral functionals, named “Koopmans functionals” with an efficient and versatile implementation of the Bethe-Salpeter equation. This methodological development will allow to address the realistic complexity of a CQD both in terms of system size and possibly functionalization via ligands at its surface.
The project is a collaborative effort of Dr. Nicola Colonna (LMS/PSI) and Dr. Nguyễn Ngọc Lĩnh (Lecturer at the Phenikaa-University, Hanoi, Vietnam)