
This year’s Nobel Prize in Chemistry goes to three researchers who have made a decisive contribution to cracking the code of proteins – important building blocks of life. However, developing applications from this knowledge, for example in medicine, requires research institutes such as PSI.
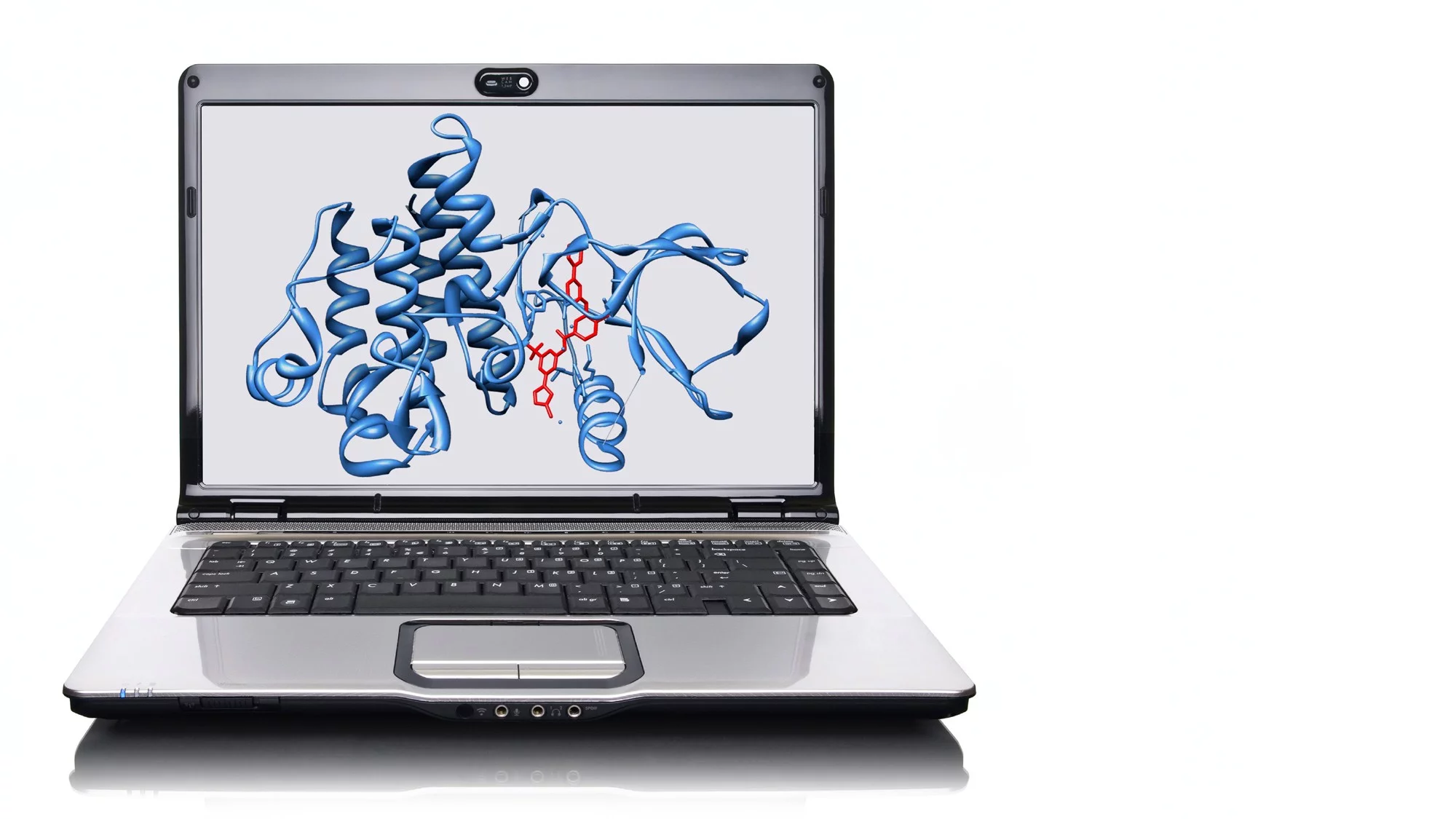
This year’s Nobel Prize in Chemistry came as a surprise in several respects. Firstly, only one of the three scientists chosen, David Baker, is a member of an academic research institution. The other two, Demis Hassabis and John Jumper, work at the Google subsidiary DeepMind. Secondly, the award is based on artificial intelligence (AI). And thirdly, the achievement being recognised draws on an Open Science project that would not have been possible without comprehensive, high-quality, open databases provided by the global scientific community – to which the Paul Scherrer Institute PSI is an important contributor. Given these unusual circumstances, it is easy to overlook the actual reason for awarding the prize. Yet that itself is revolutionary enough: The Nobel Committee is paying tribute to the three scientists for a breakthrough in protein research. Working at the company DeepMind, two of them developed an AI called AlphaFold which is able to predict the spatial structure of a protein with astonishing precision. This structure is a result of the way the molecule is folded, which in turn depends on the sequence of amino acids it contains.
Spatial folding is crucial
It is difficult to assess the full extent of the new possibilities offered by AlphaFold. Proteins and their spatial folding form the central basis of all biological systems – disrupting them can have fatal consequences. The form, function and activity of every single cell are controlled by proteins. This also holds true for the 30 trillion or more cells that make up the human body, or course, including the cells of the immune system and the brain, but also pathologically modified cancer cells. Some extra-cellular structures produced by cells are also made from proteins. These include collagen, which gives skin, bones, tendons and connective tissue their structure and strength. However, until recently scientists were often puzzled as to how the sequence of amino acids, which is relatively easy to determine, gives rise to the three-dimensional configuration.
To determine the spatial structure of proteins, which is crucial for their biological function, researchers had to resort to highly complex X-ray crystallography experiments, which often took years. Only in recent years has it become possible to achieve this by means of a particularly high-resolution form of electron microscopy. X-ray crystallography was first successfully used to determine the structure of a protein in 1959; the protein in question was myoglobin, the mussel protein which is responsible for intramuscular oxygen transport. The scientists led by Max Perutz, who was awarded the Nobel Prize for Chemistry in 1962, turned the protein into a crystal and sent monochromatic X-rays through it, similar to the radiation produced by Swiss Light Source SLS at PSI. The resulting diffraction pattern can be used to determine the folding of the protein chain – and thus provide information about the function of the protein. The location of active centres, for example, which interact with small molecules.
At the time that AlphaFold was developed, the structure of some 140,000 proteins had been determined experimentally. These are all listed in the Protein Data Bank (PDB), established in 1971, which is freely accessible to scientists and the general public. “More than five percent of the data it contains comes from the Swiss Light Source SLS at PSI,” says Jörg Standfuss, Head of the Laboratory of Biomolecular Research, which focuses on structural biology at the PSI Centre for Life Sciences. Most of the rest comes from other research centres that operate a high-quality X-ray source.
Extremely accurate predictions
The development of AlphaFold – and with it the 2024 Nobel Prize in Chemistry – would not have been possible without the huge amount of data in the PDB and its ready availability. The model combines information about the sequence of amino acids and the physical forces that govern protein folding with the experimentally determined three-dimensional structures. Using this approach, AlphaFold has predicted over 200 million structures in a very short space of time and made these available to the general public. “The accuracy of the predictions for simple proteins is now very high,” explains Standfuss. “At the moment, however, the dynamic processes that take place in proteins cannot be predicted at all, and their interactions with drugs, which are essential for the development of new types of medication, can only be predicted to a limited degree.” This is where the experimental data collected at the free-electron laser SwissFEL and the SLS at PSI is making a vital contribution to modern structural biology, with cutting-edge research in the field of structure determination of protein dynamics and the automated determination of protein structures with small chemical binding partners. Once it has been upgraded to SLS 2.0, which is due to go into operation in 2025, the facility will be able to work out experimental structures in a matter of seconds. According to Standfuss, this will provide the necessary experimental basis for the next generation of AI models.
Videos on the topic
Contact
Dr. Jörg Standfuss
Centre for Life Sciences
Paul Scherrer Institute PSI
+41 56 310 25 86
joerg.standfuss@psi.ch