
Der diesjährige Nobelpreis für Chemie geht an drei Forscher, die entscheidend dazu beigetragen haben, den Code der Proteine – wichtige Bausteine des Lebens – zu knacken. Doch damit aus diesem Wissen auch Anwendungen zum Beispiel in der Medizin entwickelt werden können, braucht es Forschungsinstitute wie das PSI.
Der Nobelpreis für Chemie überrascht dieses Jahr in mehrfacher Hinsicht. Zum einen ist von den drei ausgezeichneten Wissenschaftlern nur einer, David Baker, Angehöriger einer akademischen Forschungseinrichtung. Die anderen beiden, Demis Hassabis und John Jumper, sind Mitarbeiter von DeepMind, einer Google-Tochter. Zum anderen ist eine Künstliche Intelligenz (KI) Grundlage der Auszeichnung. Und drittens fusst die Prämierung auf einem Open Science Projekt, das ohne umfassende, hochwertige, offene Datenbanken der weltweiten wissenschaftlichen Gemeinschaft – mit dem Paul Scherrer Institut PSI als wichtigen Zulieferer – unmöglich gewesen wäre. Angesichts der ungewöhnlichen Umstände, könnte man den eigentlichen Grund der Auszeichnung beinahe übersehen. Dabei ist er für sich allein schon revolutionär genug: Das Nobelkomitee zeichnet die drei Wissenschaftler für einen Durchbruch in der Proteinforschung aus. Zwei von Ihnen entwickelten in der Firma DeepMind eine KI namens AlphaFold, welche die räumliche Struktur eines Proteins im Computer mit erstaunlicher Präzision vorhersagen kann. Dabei handelt es sich um die Art seiner Faltung, die sich auf Grund der Abfolge der enthaltenen Aminosäuren ergibt.
Räumliche Faltung entscheidend
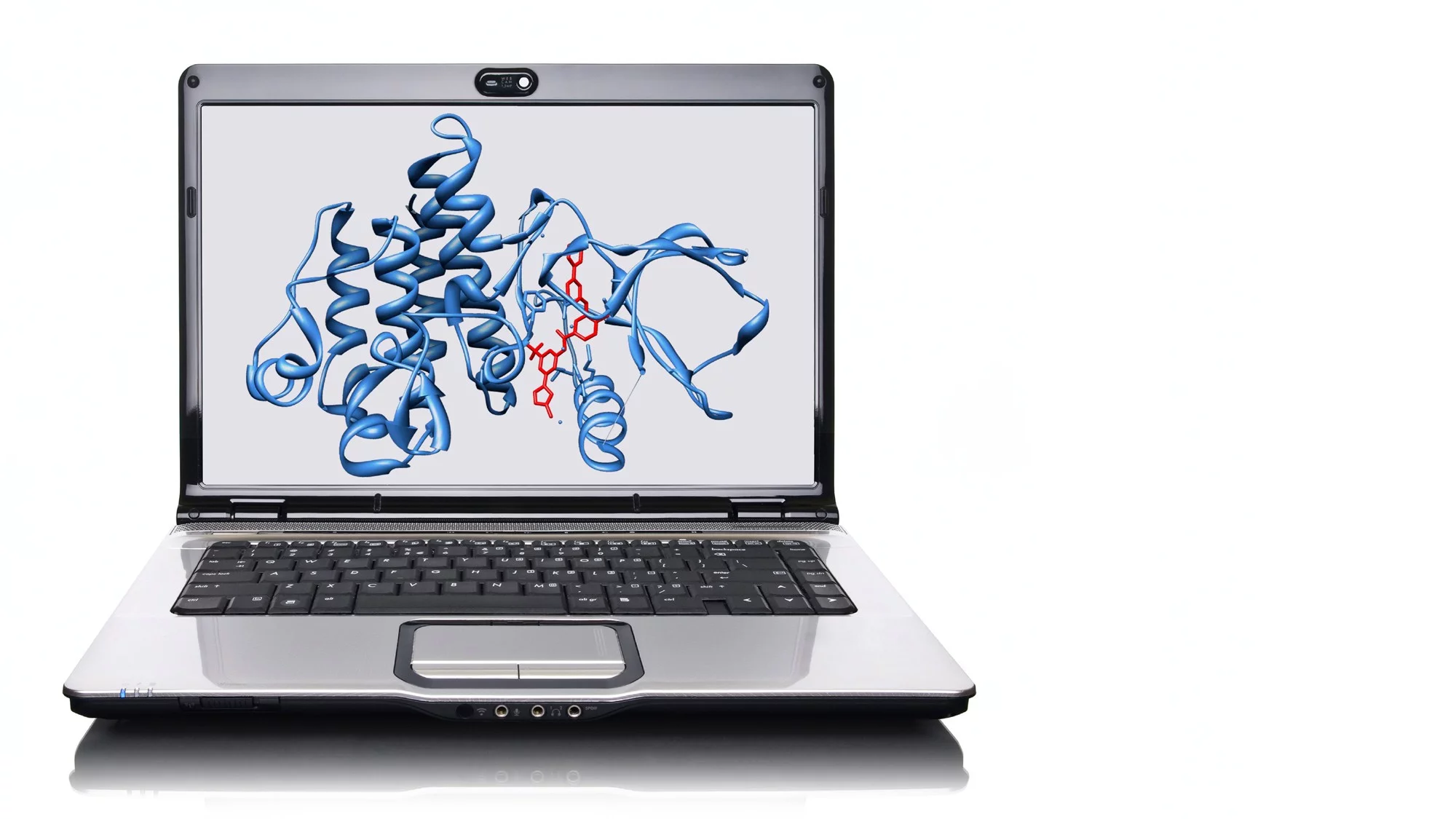
Die Tragweite der neuen Möglichkeiten durch AlphaFold ist immer noch nicht völlig abschätzbar. Proteine und ihre räumliche Faltung sind die zentrale Basis aller biologischen Systeme – ihre Störung kann entsprechend fatale Folgen haben. Die Form, Funktion und Aktivität jeder Zelle werden durch Proteine gesteuert. Das gilt selbstverständlich auch für die mehr als 30 Billionen Zellen, aus denen der menschliche Körper besteht, inklusive der Zellen des Immunsystems, des Gehirns, aber auch pathologisch veränderter Krebszellen. Auch extra-zelluläre Strukturen, die von Zellen produziert werden, bestehen aus Protein. Dazu zählt unter anderem Kollagen, das für die Struktur und Festigkeit von Haut, Knochen, Sehnen und Bindegewebe sorgt. Doch die Wissenschaft stand bis vor kurzem oft vor einem Rätsel, wie aus der leicht zu ermittelnden Abfolge der Aminosäuren seine räumliche Gestalt entsteht.
Die für die biologische Funktion entscheidende räumliche Struktur der Proteine ermittelten die Forscher in sehr aufwendigen, oft Jahre dauernden röntgenkristallographischen Experimenten. Erst seit einigen Jahren ist dies auch mit einer besonders hochauflösenden Form der Elektronenmikroskopie möglich. Die Strukturbestimmung von Proteinen mittels Röntgenkristallographie war erstmals im Jahr 1959 beim Blutfarbstoff Hämoglobin erfolgreich, dem Sauerstoff-Transporter im Körper. Die Wissenschaftler um Max Perutz, 1962 mit dem Nobelpreis für Chemie ausgezeichnet, überführten dazu das Protein in ein Kristall und bestrahlten es mit monochromatischem Röntgenlicht, wie es etwa die Synchrotron Lichtquelle Schweiz SLS am PSI zur Verfügung stellt. Aus dem Beugungsmuster des Lichts lässt sich die Art der Faltung erkennen – und damit Informationen über die Funktionsweise eines Proteins gewinnen. Zum Beispiel, wo aktive Zentren sitzen, die Interaktionen mit kleinen Molekülen (im Falle des Hämoglobin mit Sauerstoff beziehungsweise CO2) ermöglichen.
Als AlphaFold entwickelt wurde, gab es rund 140 000 experimentell ermittelte Proteinstrukturen. Sie alle sind in der 1971 gegründeten Protein Data Bank (PDB) verzeichnet, die für Wissenschaftler wie Öffentlichkeit frei zugänglich ist. «Über fünf Prozent der Daten darin stammen aus der SLS am PSI», sagt Jörg Standfuss, Leiter des Labors für biomolekulare Forschung, das im PSI Center for Life Sciences seinen Schwerpunkt auf der Strukturbiologie hat. Der Rest kommt überwiegend von anderen Forschungsstellen mit einer hochwertigen Röntgenquelle.
Sehr genaue Voraussagen
Erst mit den riesigen Datenmengen der PDB und ihrer leichten Verfügbarkeit war die Entwicklung von AlphaFold – und damit der Nobelpreis für Chemie 2024 – möglich. Das Modell kombiniert Informationen über die Abfolge der Aminosäuren sowie der physikalischen Kräfte, die die Proteinfaltung steuern, mit den experimentell ermittelten dreidimensionalen Strukturen. Dank dieser Herangehensweise hat AlphaFold über 200 Millionen Strukturen in kürzester Zeit vorhergesagt und der Allgemeinheit zur Verfügung gestellt. «Die Genauigkeit der Vorhersage bei einfachen Proteinen ist mittlerweile sehr hoch», erklärt Standfuss. «Allerdings lassen sich dynamische Prozesse in Proteinen noch gar nicht, und Interaktionen mit Wirkstoffen, die in der Medikamentenentwicklung essentiell sind, nur begrenzt vorhersagen.» Hier leisten die experimentellen Daten, die an dem Freie Eletronenlaser SwissFEL und der SLS am PSI mit Spitzenforschung im Bereich der Strukturbestimmung von Proteindynamik und der automatisierten Bestimmung von Proteinstrukturen mit kleinen chemischen Bindungspartnern gemessen werden, einen essenziellen Beitrag zur modernen Strukturbiologie. Nach ihrem Upgrade auf SLS 2.0, die 2025 eröffnet wird, werde es möglich, experimentelle Strukturen in Sekundenschnelle aufzulösen. Dies bilde die notwendige experimentelle Grundlage, so Standfuss, für die nächsten Generationen der KI-Modelle.