
A catalyst made of the noble metal ruthenium supported on a carbon substrate is frequently used industrially. A prime example is the synthesis of ammonia, which, among other things, is involved in the production of nitrogenous fertilisers. Many research groups all over the world are looking to optimise this type of catalyst as it would increase the efficiency of one of the economically most important industrial processes. However, our understanding of how the catalytically active centres in the catalyst develop has been somewhat patchy thus far. Researchers from the Paul Scherrer Institute PSI can now unveil some fresh insights.

Ruthenium belongs to the platinum metal family and, like platinum itself, is thus a popular component of catalysts, which are used to kick-start chemical reactions that would otherwise eventually grind to a standstill. The catalytic effect of ruthenium is used in the Haber-Bosch process, for instance, where ammonia and subsequently valuable fertilisers are synthesised from nitrogen and hydrogen for agriculture. Sometimes, a ruthenium catalyst is also deployed in methanation, namely the production of methane (synthetic natural gas) from hydrogen and carbon monoxide or dioxide. In each of these catalytic reactions, it all boils down to maximising the number of catalytically active centres in the catalyst – the only way to exploit the full potential of the expensive noble metal.
What counts: steps and terraces
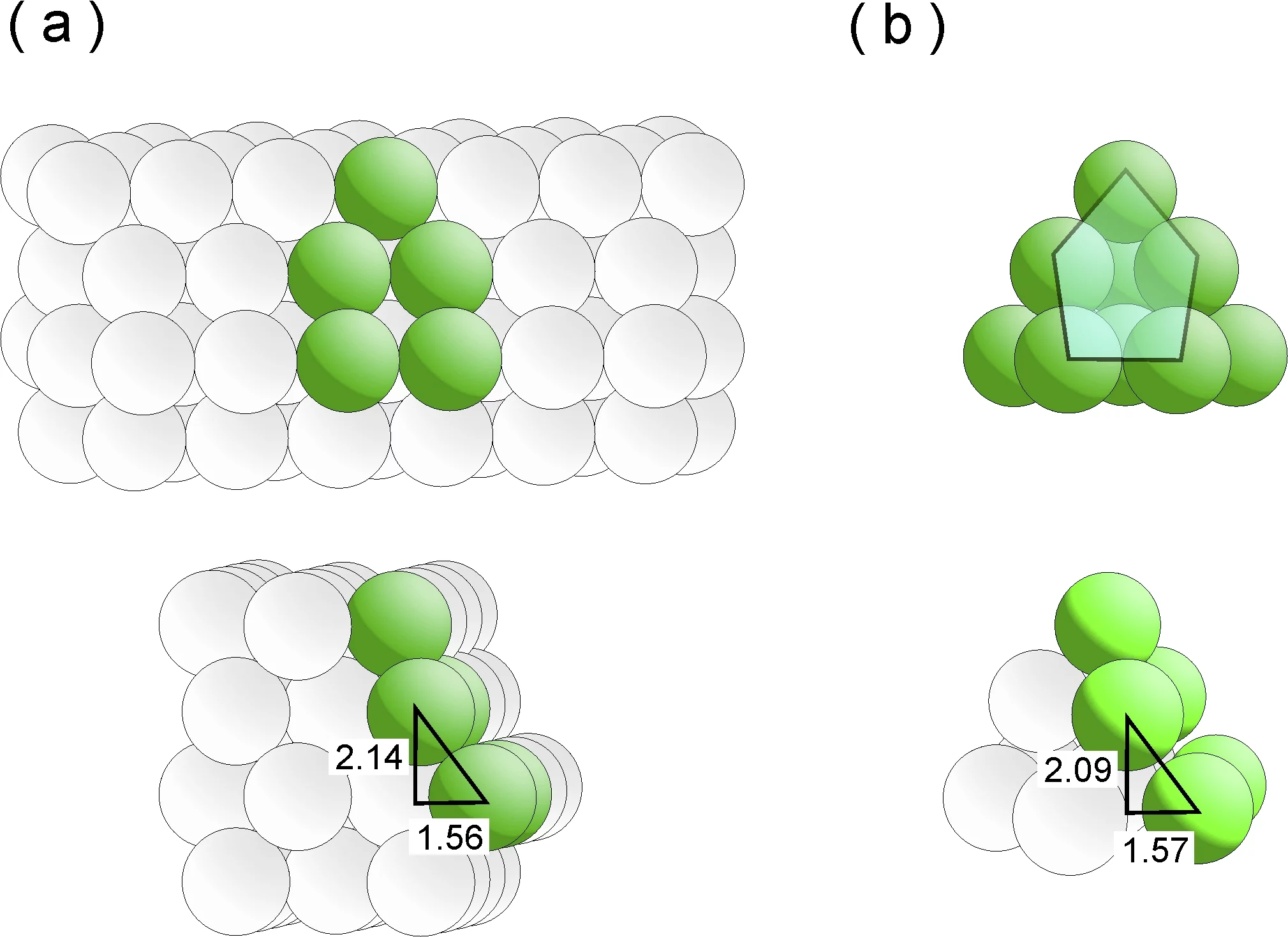
The researchers have known one thing for some time: good catalysts are rough and ready, not smooth – an otherwise hackneyed description that is particularly apt in the microscopic world of catalysis. After all, an efficient catalyst essentially has two functions: firstly, to “lure” the reactive molecule into attaching itself to its surface, and secondly to keep these adsorbates under its spell for long enough to trigger the desired reaction. For this to work, however, the surface of the catalyst needs to have imperfect points, such as steps and terraces, as the molecules would not have a favourite place to settle upon on a perfectly smooth, uniform surface and would “hop” from one point to the next as a result. In the case of the ruthenium catalyst, the particularly attractive docking points are referred to as B5 sites. The researchers Izabela Czekaj and Jörg Wambach from the PSI have now, with the aid of complex calculations, succeeded in recreating how B5 sites might form by simulating exactly the growth of the ruthenium particles on the carbon substrate.
B5 sites are three-dimensional structures that comprise five ruthenium atoms and are arranged on two distinct planes. The three atoms on the lower plane form a terrace-like area, the two atoms on the upper tier a step to it. The structure effectively looks like an unfolded deckchair. Molecules especially like docking onto the step, as if drawn by a magnet. In thermodynamics jargon, the system’s energy is said to be minimised.
Previous models too inaccurate
Researchers have puzzled long and hard over exactly how the B5 sites are formed. Previous simulations that were supposed to help understand the structure formations were evidently based on over-simplified assumptions and coarse approximations. For instance, the influence of the carbon substrate was either underestimated or ignored altogether. And this is precisely where the PSI researchers came in. With the aid of density functional theory (DFT), a method of calculation that derives the details of structure formation directly from fundamental laws, they were able to work out the growth of the ruthenium particles on different carbon substrates with unprecedented precision. “DFT calculations of nanometre-sized metal particles on a substrate are complex and time-consuming and Izabela Czekaj is one of the few scientists worldwide to have mastered this technique,” says Wambach.
DFT calculations produce better image
In order to verify whether their DFT calculations actually produce a faithful image of the ruthenium catalyst, the PSI researchers used DFT calculations to simulate on the computer how a Ru catalyst would absorb x-ray radiation and how x-ray radiation would kick off free electrons from the catalyst’s inner structure. The corresponding spectra – referred to as x-ray absorption and photoelectron emission spectra – were then measured on an industrially manufactured Ru catalyst and compared with the simulated spectra. The close agreement between the simulations and the experiment revealed that the DFT calculations yield an extremely sharp image of the catalyst.
“By analysing the fine structure of the x-ray absorption spectra, we can infer some parameters related to the three-dimensional structure of the catalyst,” explains Wambach. To do so, however, you need models that describe this very structure to begin with. The problem is that previous models proved inadequate as they were unable to reproduce the measured x-ray absorption spectra. The PSI researchers’ density functional calculations provide structural data that produces considerably better results when evaluating the x-ray absorption spectra – for Wambach, a clear indication that the detailed DFT calculations, although time-consuming, have one major advantage: they describe the reality of the three-dimensional structure in the catalyst more effectively than other models usually applied.
“Catalysis research always means a quest for a better understanding of the relation between the three-dimensional structure and reactivity,” says Wambach. However, the application of the DFT method cannot just describe the experiments more effectively; it also gives the researchers the possibility of examining the conditions under which the maximum number of B5 sites form on a ruthenium catalyst as well as the influence of the substrate on the structure. This is an initial step towards the distant goal of eventually producing such catalysts at will.
Text:Leonid Leiva
Contact
Dr. Jörg Wambach, Bioenergy and Catalysis Laboratory, Paul Scherrer Institute,Telephone: +41 56 310 4266, E-Mail: joerg.wambach@psi.ch