
Le catalyseur au ruthénium sur support de carbone est couramment utilisé dans l’industrie. Exemple éminent : la synthèse d’ammoniac, qui sert notamment à la fabrication d’engrais azotés. L’objectif de nombreux groupes de chercheurs dans le monde est d’optimiser ce type de catalyseur, dans l’idée d’améliorer l’efficacité d’un procédé industriel, qui figure parmi les plus importants d’un point de vue économique. Mais jusqu’ici, les connaissances sur la manière dont se constituent les centres catalytiquement actifs dans le catalyseur restent lacunaires. Des chercheurs de l’Institut Paul Scherrer (PSI) ont à présent réussi à mettre à jour des éléments importants.

Le ruthénium est un métal noble du groupe du platine, et à l’instar de ce dernier, c’est un composant apprécié des catalyseurs. Ces substances permettent d’accélérer la vitesse de réactions chimiques, qui sinon traîneraient en longueur. L’effet catalytique du ruthénium intervient par exemple dans le cadre du procédé Haber-Bosch, lors duquel sont synthétisés de l’ammoniac, et par la suite de précieux engrais pour l’agriculture, à partir d’azote et d’hydrogène. Lors de la méthanisation également, c’est-à-dire la méthanation (gaz artificiel) à partir d’hydrogène et de monoxyde de carbone, respectivement de dioxyde de carbone, on utilise parfois un catalyseur au ruthénium. Pendant toutes ces réactions catalytiques, il s’agit de maximiser le nombre de centres catalytiquement actifs dans le catalyseur. C’est ainsi seulement que l’on peut exploiter au maximum le plein potentiel du coûteux métal noble.
Ce qui compte : les aspérités
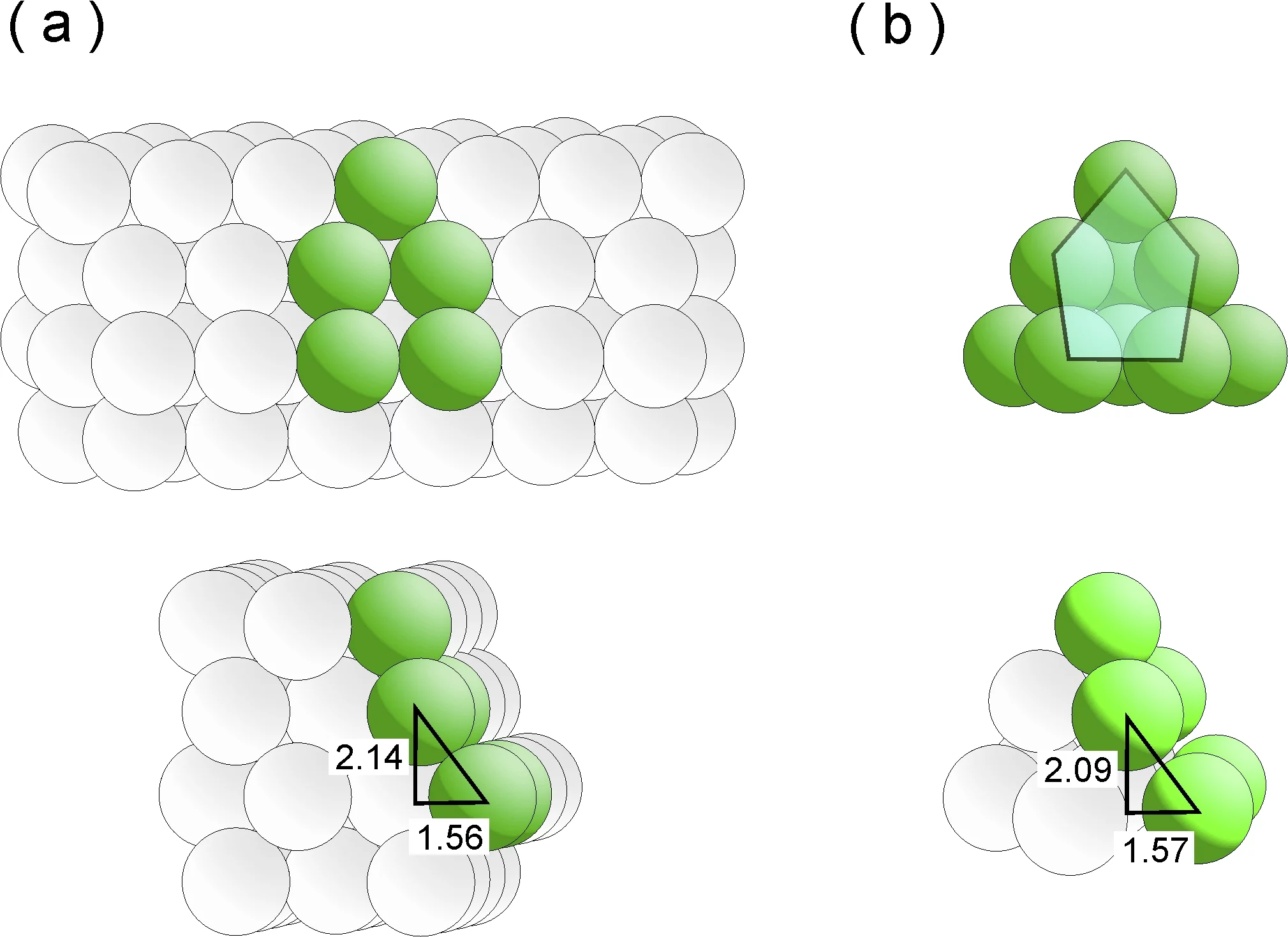
Les chercheurs savent depuis longtemps que pour être bons, les catalyseurs ne doivent surtout pas être lisses, mais dotés d’aspérités. Dans l’univers microscopique de la catalyse, cette description n’est pas seulement métaphorique. Fondamentalement, un catalyseur efficace doit en effet assumer deux tâches : premièrement, il doit « séduire » les molécules qui réagissent pour que ces dernières viennent s’arrimer à sa surface. Et deuxièmement, il doit captiver ces adsorbats suffisamment longtemps pour que la réaction souhaitée se produise. Mais pour que ce soit possible, il faut que la surface du catalyseur présente des imperfections, comme des marches ou des terrasses, car sur une surface parfaitement lisse et uniforme, les molécules n’auraient pas de sites de prédilection où s’installer. En conséquence de quoi, elles « sautilleraient » sans cesse d’un site à l’autre. Dans un catalyseur au ruthénium, les points d’arrimage particulièrement attrayants sont appelés sites B5. Izabela Czekaj et Jörg Wambach, chercheurs au PSI, ont à présent reproduit, à l’aide de calculs complexes, la façon dont les sites B5 peuvent se constituer. Ils y sont parvenus en simulant avec précision la croissance de particules de ruthénium sur un substrat de carbone.
Les sites B5 sont des structures tridimensionnelles, composées de cinq atomes de ruthénium, répartis sur deux niveaux. Les trois atomes du niveau inférieur forment une surface en terrasse, et les deux atomes du niveau supérieur une marche vers cette terrasse. Dans l’ensemble la structure ressemble à une chaise longue inclinée vers l’arrière. Les molécules s’arriment particulièrement volontiers aux atomes de la marche, qui les attirent quasiment comme un aimant. Dans le langage de la thermodynamique, on parle de minimisation de l’énergie du système.
Jusqu’ici, des modèles trop imprécis
Mais la formation des sites B5 reste à ce jour une énigme pour les chercheurs. Les simulations, mises au point jusqu’ici pour comprendre la formation des structures, étaient apparemment fondées sur des simplifications et des approximations grossières. Ainsi, l’influence du substrat de carbone était sous-estimée, voire complètement négligée. C’est précisément là-dessus que se sont concentrés les chercheurs du PSI. A l’aide de la théorie de la fonctionnelle de la densité (DFT), une méthode de calcul qui déduit directement la formation d’une structure à partir de lois fondamentales, ils ont réussi à calculer la croissance des particules de ruthénium sur différents substrats de carbone avec une précision inégalée à ce jour. « Les calculs DFT de particules métalliques de l’ordre du nanomètre sur un support sont complexes et longues, souligne Jörg Wambach. Izabela Czekaj est l’une des rares scientifiques au monde qui les maîtrisent. »
Des calculs DFT fournissent une meilleure image
Pour vérifier si les calculs DFT fournissaient bel et bien une image véridique du catalyseur au ruthénium, les chercheurs du PSI ont simulé par ordinateur, sur la base de calculs DFT, la façon dont un catalyseur Ru absorberait les rayons X, respectivement la façon dont ces rayons X arracheraient des électrons au catalyseur. Les spectres correspondants – appelés spectres d’absorption des rayons X et spectres d’émission des photoélectrons – ont ensuite été mesurés sur un catalyseur Ru de fabrication industrielle. La cohérence entre simulations et expérience a montré que les calculs DFT fournissaient une image très nette du catalyseur. « L’analyse de la structure fine des spectres d’absorption des rayons X permet des déductions sur la structure tridimensionnelle du catalyseur », explique Jörg Wambach. Mais pour ce faire, il faut partir de modèles qui décrivent précisément cette structure. Or, les modèles disponibles se sont avérés défectueux, et incapables de reproduire les spectres d’absorption des rayons X mesurés. Avec les calculs de la fonctionnelle de la densité des chercheurs du PSI, on obtient des données structurelles, qui fournissent des résultats bien meilleurs lors de l’évaluation des spectres d’absorption des rayons X. Pour Jörg Wambach, cela constitue un indice clair de l’avantage décisif que présentent les calculs DFT détaillés, en dépit du temps qu’ils nécessitent : ils décrivent mieux la réalité de la structure tridimensionnelle dans le catalyseur que les modèles couramment utilisés.
« Faire de la recherche sur les catalyseurs, c’est comprendre toujours mieux la relation de cause à effet entre structure tridimensionnelle et réactivité », rappelle Jörg Wambach. Mais l’application de la méthode DFT ne permet pas seulement de mieux décrire les expériences : elle fournit aussi aux chercheurs la possibilité d’étudier les conditions dans lesquelles le nombre maximum de sites B5 se constitue sur un catalyseur au ruthénium, et l’influence du matériau porteur sur la structure. C’est un premier pas vers un objectif lointain : pouvoir un jour fabriquer ce genre de catalyseur, à la demande.
Auteur: Leonid Leiva
Contact
Dr. Jörg Wambach, Laboratoire Bioénergie et Catalyse , Institut Paul Scherrer,Téléphone: +41 56 310 4266, E-Mail: joerg.wambach@psi.ch