
Ein Katalysator aus dem Edelmetall Ruthenium auf einem Kohlenstoffträger wird häufig industriell eingesetzt. Ein prominentes Beispiel ist die Synthese von Ammoniak, welches unter anderem zur Herstellung von stickstoffhaltigen Düngemitteln dient. Diesen Katalysatortyp zu optimieren ist das Ziel vieler Forschungsgruppen weltweit, würde dies doch die Effizienz eines der ökonomisch bedeutendsten Industrieprozesse erhöhen. Doch das Verständnis dessen, wie es zum Aufbau der katalytisch aktiven Zentren im Katalysator kommt, ist bisher lückenhaft. Forscher des Paul Scherrer Instituts PSI bringen nun ein paar wichtige Erkenntnisse ans Licht.

Ruthenium gehört zur Familie der Platinmetalle und ist somit wie Platin selbst ein beliebter Bestandteil von Katalysatoren, mit denen sich sonst träge vor sich hin dümpelnde chemische Reaktionen auf Trab bringen lassen. Die katalytische Wirkung von Ruthenium kommt etwa beim Haber-Bosch-Prozess zum Einsatz, bei dem aus Stickstoff und Wasserstoff Ammoniak und in der Folge wertvolle Düngemittel für die Landwirtschaft synthetisiert werden. Auch bei der Methanisierung, das heisst der Herstellung von Methan (künstlichem Erdgas) aus Wasserstoff und Kohlenmonoxid bzw. Kohlendioxid, wird bisweilen auf einen Rutheniumkatalysator gesetzt. Bei all diesen katalytischen Reaktionen kommt es darauf an, die Anzahl der katalytisch aktiven Zentren im Katalysator zu maximieren. Nur so kann man aus dem teuren Edelmetall das volle Potenzial schöpfen.
Was zählt: Ecken und Kanten
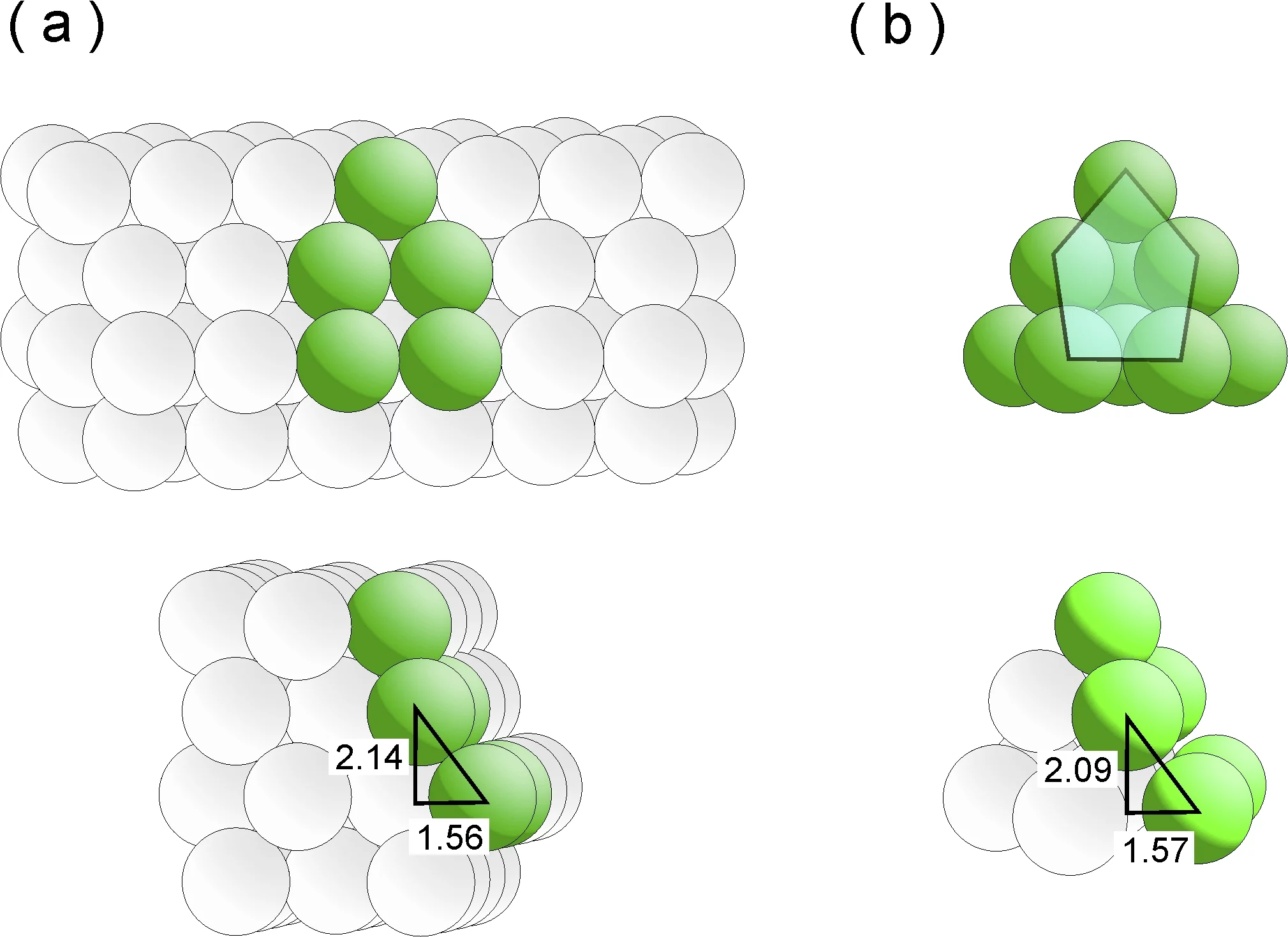
Eines wissen die Forscher schon lange: gute Katalysatoren sind keine aalglatten Zeitgenossen, sondern vielmehr mit Ecken und Kanten ausgestattet. In der mikroskopischen Welt der Katalyse trifft diese sonst überstrapazierte Beschreibung buchstäblich zu. Denn ein effizienter Katalysator muss im Grunde zweierlei bewältigen: Erstens muss er die reagierenden Moleküle zum Andocken an seine Oberfläche „verführen“. Und zweitens muss er diese Adsorbate auch lange genug in seinem Bann halten, so dass es zur gewünschten Reaktion kommt. Dazu muss die Oberfläche des Katalysators aber imperfekte Stellen wie Stufen oder Terassen aufweisen, denn auf einer perfekt glatten, immer gleich aussehenden Oberfläche hätten die Moleküle keine bevorzugten Stellen, um sich niederzulassen. Sie würden folglich immerzu von einer Stelle zur anderen „herumspringen“. Die beim Rutheniumkatalysator besonders attraktiven Andockstellen werden als B5-Stellen bezeichnet. Wie B5-Stellen zustande kommen könnten, haben nun die Forschenden Izabela Czekaj und Jörg Wambach vom PSI mit Hilfe von aufwändigen Berechnungen nachgebildet. Dies gelang ihnen, indem sie das Wachstum der Ruthenium-Partikeln auf dem Kohlenstoffsubstrat exakt simulierten.
B5-Stellen sind dreidimensionale Strukturen, die aus fünf Rutheniumatomen bestehen und über zwei Ebenen verteilt sind. Die drei Atome der unteren Ebene bilden eine terassenartige Fläche, die zwei Atome der oberen Ebene bilden zu dieser eine Stufe. Insgesamt mutet die Struktur wie ein zurückgelehnter Liegestuhl an. An den Atomen der Stufe docken Moleküle besonders gerne an, sie werden quasi wie von einem Magnet angezogen. In der Sprache der Thermodynamik sagt man, dass dann die Energie des Systems minimiert wird.
Bisherige Modelle zu ungenau
Doch wie es genau zur Bildung von B5-Stellen kommt, darüber rätselten Forscher bisher weitgehend. Bisherige Simulationen, mit denen die Strukturbildungen nachvollzogen werden sollte, gründeten offenbar auf zu groben Vereinfachungen und Näherungen. So wurde zum Beispiel der Einfluss des Kohlenstoffsubstrats entweder unterschätzt oder ganz vernachlässigt. Und genau hier setzten die PSI-Forscher an. Mit Hilfe der Dichtefunktionaltheorie (DFT), einer Berechnungsmethode, die die Strukturbildung direkt aus fundamentalen Gesetzen ableitet, konnten sie das Wachstum der Rutheniumpartikeln auf unterschiedlichen Kohlenstoff-Substraten mit noch nie erreichter Präzision berechnen. „DFT-Berechnungen von nanometergrossen Metallpartikeln auf einem Träger sind komplex und zeitaufwändig und Izabela Czekaj ist eine der wenigen Wissenschaftlerinnen weltweit, die sie beherrscht“, sagt Wambach.
DFT-Berechnungen liefern ein besseres Bild
Um zu überprüfen, ob ihre DFT-Berechnungen tatsächlich ein wahrheitsgetreues Bild des Rutheniumkatalysators liefern, simulierten die PSI-Forschenden am Computer auf der Basis von DFT-Berechnungen, wie ein Ru-Katalysator Röntgenstrahlung absorbieren bzw. wie die Röntgenstrahlung Elektronen aus dem Katalysator herausschlagen würde. Die entsprechenden Spektren -Röntgenabsorptions- und Photoelektronenemissionsspektren genannt- wurden dann an einem industriell hergestellten Ru-Katalysator gemessen und mit den simulierten Spektren verglichen. Die gute Übereinstimmung zwischen Simulationen und Experiment zeigte, dass die DFT-Berechnungen ein sehr scharfes Bild des Katalysators liefern.
„Indem man die Feinstruktur der Röntgenabsorptionsspektren analysiert, lassen sich Rückschlüsse auf die dreidimensionale Struktur des Katalysators ableiten“, erläutert Wambach. Dazu muss man aber von Modellen ausgehen, die ebendiese Struktur beschreiben. Nur, die bisherigen Modelle erwiesen sich als mangelhaft: sie konnten die gemessenen Röntgenabsorptionsspektren nicht reproduzieren. Mit den Dichtefunktionalberechnungen der PSI-Forscher erhält man Strukturdaten, die beim Auswerten der Röntgenabssorptionspektren deutlich bessere Resultate liefern. Das ist für Wambach ein klarer Hinweis darauf, dass die detaillierten DFT-Berechnungen, obwohl zeitaufwändig, einen entscheidenen Vorteil haben: sie beschreiben die Realität der dreidimensionalen Struktur im Katalysator besser als sonst üblicherweise angewandte Modelle.
„Katalyseforschung bedeutet stets, den Zusammenhang von dreidimensionaler Struktur und der Reaktivität besser zu verstehen“, sagt Wambach. Die Anwendung der DFT-Methode kann aber nicht nur die Experimente besser beschreiben: sie gibt den Forschern auch die Möglichkeit, zu untersuchen, unter welchen Bedingungen auf einem Ruthenium-Katalysator die maximale Anzahl von B5-Stellen entsteht und welchen Einfluss das Trägermaterial auf die Struktur hat. Dies ist ein erster Schritt hin zum fernen Ziel, solche Katalysatoren eines Tages nach Wunsch herzustellen.
Text: Leonid Leiva
Kontakt / Ansprechpartner
Dr. Jörg Wambach, Labor für Bioenergie und Katalyse, Paul Scherrer Institut,Telefon: +41 56 310 4266, E-Mail: joerg.wambach@psi.ch